The muscle atonia that
characterizes REM sleep results from the hyperpolarization
of the spinal cord’s motor neurons by glycine, an inhibitory
neurotransmitter released by neurons in the brainstem. Researchers
have found, however, that the motor nuclei of the cranial
nerves are not strongly inhibited by glycine, which would
explain why movements of eye and facial muscles persist during
REM sleep.
When you are awake,
your brain’s wakefulness circuits exert controls that
prevent you from displaying the forms of brain activity that
characterize REM sleep. But in the human fetus, these controls
are not yet in place, which may explain why, during the last
few months of gestation, the fetus spends such a large proportion
of its sleeping time (about 80%) in REM sleep.
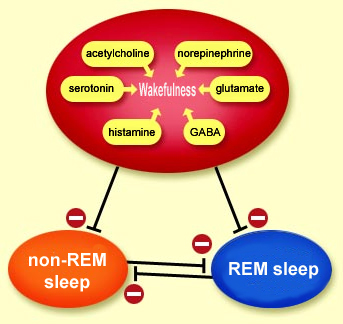
THE NEURONAL SWITCHES FOR WAKING AND SLEEPING
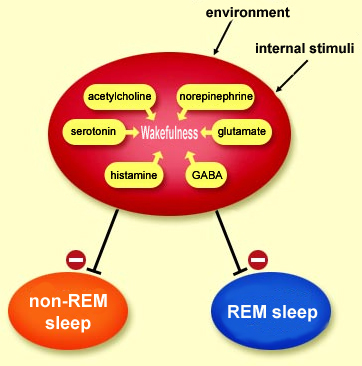
Activity
in the brain’s wakefulness circuits, promoted
by stimuli from the internal and external environments,
prevents the onset of sleep. These neuronal wakefulness
circuits can thus be described as an inhibitory permissive
system for the two types of sleep, non-REM and REM. Only
when the inhibition imposed by this system is lifted can
the brain go through the alternating
periods of non-REM sleep and REM sleep, which involve
diametrically opposite metabolic states. In non-REM sleep,
the basal metabolism is lowered to save energy. In REM
sleep, the metabolism is high–just as high as when
the individual is awake.
Except in the presence of certain pathologies such as narcolepsy,
the first period of sleep is always non-REM sleep, and its purpose
is often regarded as being to prepare for REM sleep. This period
of non-REM sleep begins with the disappearance of the cholinergic
effects of wakefulness, which disinhibits
the “pacemaker” neurons of the thalamic reticular nucleus.
These neurons then impart their rhythm to the thalamocortical neurons,
which then in turn induce their “slow waves” throughout
the cortex.
As non-REM
sleep grows deeper, the aminergic neuromodulation exerted by the neurons
of the locus
coeruleus and the raphe nucleus also subsides gradually, preparing the brain
for REM sleep. During the first stages of non-REM sleep, the discharge rate of
these noradrenergic and serotonergic neurons decreases slightly, but during the
transition from non-REM sleep to REM sleep, it decreases dramatically, until
these cells almost completely stop firing.
The cellular mechanisms
of REM sleep are complex, because its onset is inhibited
not only by the permissive system of the wakefulness circuits
as just described, but also by certain systems that come
into play during non-REM sleep. Be that as it may, the removal
of aminergic inhibition combined with the effects of other
factors can be said to release the “executive mechanisms” for
sleep, in which acetylcholine plays a central role.
The neuronal populations associated with wakefulness,
non-REM sleep, and REM sleep thus act somewhat like switches
for one another: one of these states becomes active when
another ceases to be active, and vice versa.
For example, such switch mechanisms control
the two totally different modes of brain function that REM sleep
and wakefulness constitute. In these two modes, the brain’s
activity is similar in all respects, except in certain specific
populations of neurons that make all the difference. It is convenient
to describe these populations as being “on”
(active) or “off” (inactive) during the various states
of wakefulness and sleep.
Other, cholinergic neurons in the medulla oblongata and the pons
that become very active during REM sleep but are inactive during
waking periods are called “REM-on”
or “wake-off”. Various groups of “REM-on” neurons
have been identified for each of the intrinsic
features of REM sleep. Taken as a whole, these various groups
of neurons constitute what is called the “executive system
for REM sleep”.
For example, the muscle atonia characteristic
of REM sleep (see sidebar) results not from a passive relaxation
of the muscles, but rather from the blocking
of the spinal motor neurons by other neurons that produce glycine.
These latter neurons are thus categorized as “REM-on”,
as are the glutamatergic neurons that project to the cholinergic
neurons, to the GABAergic neurons of the posterior hypothalamus,
to the oculomotor nuclei (the eyes move during REM sleep), and
to the medullary reticular formation responsible for muscle paralysis.
Lastly, two other groups of “REM-on” neurons
play a key role in REM sleep. The first group, in the oral
pontine reticular nucleus , show little or no activity during
waking periods and non-REM sleep but are very active during REM
sleep. The second group consists of certain GABAergic neurons that
inhibit serotonergic and noradrenergic activity during REM sleep.
No more than about 50 minutes after a period
of REM sleep begins, the REM-off neurons become active, releasing
norepinephrine and serotonin. By countering the effects of acetylcholine,
these neurotransmitters switch off the intense activity associated
with REM sleep, and this period of REM sleep comes to an end.
The role of serotonin is especially complex,
because this neurotransmitter is also directly involved in the process
of falling asleep.
The dendrites of the
pyramidal neurons in the cortex receive many connections
from other neurons. When the afferent axons from these
other neurons are activated, their terminals release neurotransmitters
into their synapses with the dendrites on the pyramidal
neurons. Some of these neurotransmitters excite the pyramidal
neurons, while others inhibit them. When an excitatory
neurotransmitter such as glutamate has
been released, it binds to the postsynaptic
receptors for glutamate on the pyramidal
neuron’s dendrite, causing channels to open in its
cell membrane and let positively
charged ions flow into the dendrite. This
positive inflow makes the extracellular environment slightly
negative. The excitatory postsynaptic potential then propagates
down the dendrite to the pyramidal neuron’s cell
body, creating a flow of ions that generates another weak
electrical current, this one running parallel to the surface
of the cell membrane rather than passing through it.
It is the sum of the currents generated
more or less synchronously by thousands of cortical neurons
that is detected by the electroencephalograph electrode
attached to a subject’s scalp. The electroencephalograph
then compares this sum to that detected by a second electrode
placed a certain distance away on the scalp. By convention,
a more negative current is represented by an upward deflection
in the EEG trace.
These rhythmic oscillations are determined in part by the
activity of the neurons of the thalamus and the feedback loops
that these neurons maintain with the cortex (see sidebar).
Each of these thalamic neurons spontaneously maintains its
own rhythmic activity and can hence be regarded as a single-neuron
oscillator. Through a particular set of membrane-potential-dependent
ion channels, these cells can send out action
potentials at a certain rhythm without having to receive
any external stimulus to do so.
But how do the spontaneous, rhythmic action potentials from
these thalamic neurons act as a powerful pacemaker for the
entire cortex? The answer is that these neurons synchronize
their activity through association
mechanisms similar to those used by multineuronal oscillators.
Thus, through their rich axonal projections to the cortex,
a relatively small group of thalamic neurons can cause a far
larger group of cortical neurons to oscillate at the same frequency
as the thalamic neurons. Researchers have found that damage
to the thalamus can reduce these cortical oscillations, or
eliminate them completely.
Adapted from Neurosciences,
Purves, Augustine, Fitzpatrick, Katz, LaMantia, McNamara,
Williams, and De Boeck, Eds., 2003
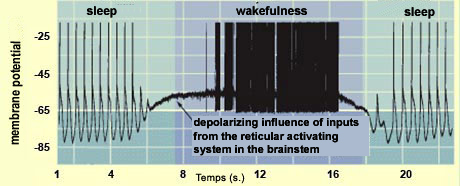
Recording
of the activity of a cortical neuron, with the oscillating
pattern characterizing sleep and the tonic activity characterizing
wakefulness.
The thalamic neurons that project their axons to the cortex
have another important electrophysiological property as well:
they can toggle between two steady states. One is the spontaneous
oscillatory activity just described, which is intrinsic
to them; the other is the tonic activity that
occurs when these neurons are depolarized by external inputs.
Conversely, hyperpolarization of the thalamocortical
neurons stabilizes their oscillatory state. This hyperpolarization
is induced by inhibitory synaptic inputs to these neurons from
the GABAergic neurons of the thalamic reticular nucleus.
These GABAergic neurons receive projections from both the brainstem
and the cortex. When these neurons transmit action potentials,
they hyperpolarize the thalamocortical neurons, which then
enter their oscillating state.
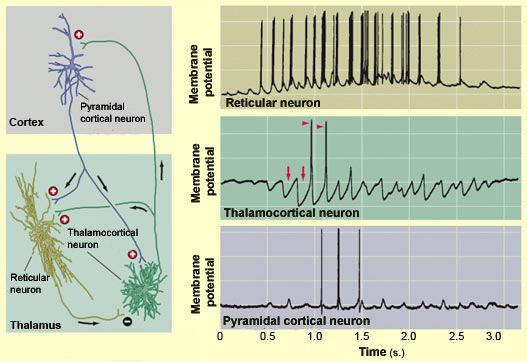
During a sleep
spindle (a phenomenon observed mainly during Stage 2 non-REM
sleep), the series of impulses transmitted by the GABAergic
neurons of the thalamic reticular nucleus hyperpolarize (red
arrows) the thalamocortical neurons and cause them to generate
action potentials (red triangles). These action potentials
are transmitted to the pyramidal neurons in the cortex, where
their summation produces the
sleep spindles recorded in the EEG.
Adapted from Neurosciences,
Purves, Augustine, Fitzpatrick, Katz, LaMantia, McNamara,
Williams, and De Boeck, Eds., 2003
When the thalamocortical neurons are
in this oscillating state, they cause the cortical neurons
to become synchronized with them, thus causing a disconnection
between the cortex and the outside world.
This disconnection is of course greatest during Stage
4 non-REM sleep, when the frequency of the EEG trace is
lowest and its amplitude is highest. These are also the times
of night when it is hardest to wake someone up.