|
|
| Funding for this site is provided by readers like you. | |
|
|
|
|
|
|||||
|
|
|||||||
|
|
|
|



|
|
Free Radicals and Aging: More Complicated Than We Thought
|
|
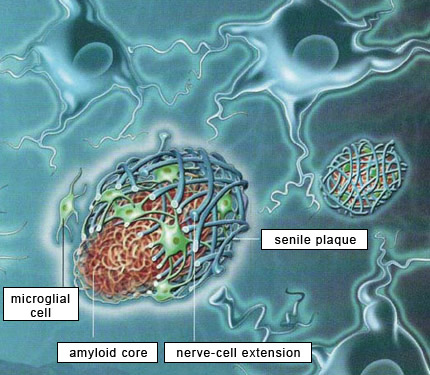
Research on Alzheimer’s has been largely dominated by what was initially known simply as the amyloid hypothesis and is now more specifically described as the beta-amyloid synaptic hypothesis. According to this hypothesis, the abnormal accumulation of beta-amyloid protein, in the form of amyloid plaques or oligomers, in the brain, is the primary mechanism that causes Alzheimer’s. The majority of experimental vaccines and medications therefore target these amyloid plaques. Within the Alzheimer’s research community, scientists whose research is based on this hypothesis are known as “BAPtists” (where BAP stands for beta-amyloid peptide). The primary opposing camp are the TAUists, who instead believe that Alzheimer’s disease develops as the result of the appearance of neurofibrillary tangles.
But the TAUists are not the only scientists who question the amyloid hypothesis. More and more researchers are beginning to voice alternative explanations. To justify their explorations off the beaten path, these scientists point out that the quarter-century history of research based on the amyloid hypothesis is littered with failures, with little tangible progress to show for it. Here is a brief summary of the flaws that these researchers see in this hypothesis. First, very little correlation has been observed between the extent of cognitive deficits in Alzheimer’s patients and the quantity of amyloid plaques in their brains. This represents a major anomaly in this paradigm that has dominated Alzheimer’s research since the start of the 1990s, if not longer. The second perceived flaw is that amyloid plaques are also found in the brains of normal individuals, something that the defenders of the amyloid hypothesis are hard-pressed to explain. In fact, “normal” brains can sometimes contain more amyloid plaques than the brains of patients with severe cases of Alzheimer’s. Hence Whitehouse and other authors have argued that the criteria for diagnosing “Alzheimer’s disease” are too vague and that Alzheimer’s cases may be nothing more than special cases of normal aging. Third, the relations between beta-amyloid protein and tau protein are complex, to say the least. Some authors say that people with Alzheimer’s-type dementia actually have two kinds of cortical pathologies—beta-amyloid and tau—simultaneously, that there is a synergy between the two, and that there is therefore no need to choose between the BAPtist and the TAUist camps. And there are in fact some data to support this view. For example, a transgenic mouse that overexpresses both beta-amyloid protein and tau protein will produce more neurofibrillary tangles than a transgenic mouse that has only the tau-protein mutation. But the overall picture remains unclear. For example, normal people can have only moderate levels of beta-amyloid deposits but at the same time have fairly extensive neurofibrillary tangles, advancing all the way into the temporal cortex in stage 6 or 7 tau pathology. And here again, cases have been reported of patients who had up to stage 6 tau pathology but no beta-amyloid deposits. Moreover, individuals age 75 and over always have some tau pathology in their transentorhinal and entorhinal cortex. But this pathology is often quite limited, and there are even people in their 90s who display very little tau pathology. This shows once again that the development of this pathology is not correlated with age in any linear, systematic way, although age is a major risk factor for tau pathology. Many researchers in fact believe that accumulations of proteins are generally only the final manifestations of diseases with earlier causes, and that amyloid plaques and neurofibrillary tangles are no exception to this rule. Some researchers even directly question the harmfulness of amyloid plaques and neurofibrillary tangles, arguing that they may in fact represent a defensive response by the brain to harmful processes that precede them, such as oxidative stress, inflammation, and dysfunctions in the cellular cycle. Thus, in contrast to the assumption, under the amyloid hypothesis, that beta-amyloid protein has no particular physiological function, many studies now report that beta-amyloid binds to specific receptors or can induce inflammatory reactions. For example, in vivo studies have shown that this protein plays a protective role against microbes. But even though beta-amyloid may contribute to a normal, helpful immune response in the short term, its prolonged activity might still have pathological consequences.
And what about the protective antioxidant properties that 40-amino-acid beta-amyloid has been shown to have? This finding, like many others, argues against the oxidative damage of Alzheimer’s that is attributed to beta-amyloid in the amyloid hypothesis (see box below). In any case, all of these “abnormal” data (in the Kuhnian sense) tend to weaken the amyloid hypothesis, which some authors say had begun to take on the trappings of dogma. Because dogmatism is a bad idea in general, and a bad idea in science in particular, many researchers are rather pleased to see the growing number of alternative hypotheses to help us better understand and treat the condition known as Alzheimer’s. These researchers also hope that research-funding agencies and scientific journals will do more to encourage the emergence of such hypotheses—something that they have not always done in the past. Beyond its academic sociological interest, this is a major societal issue, considering the resources invested in developing treatments based on the amyloid hypothesis and the hopes that such treatments arouse in people with Alzheimer’s and their families.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


| |
|
|
|
|
|
|
|
|