|
|
| Funding for this site is provided by readers like you. | |
|
|
|
|
|
|||||
|
|
|||||||
|
|
|
|


|
|
| |
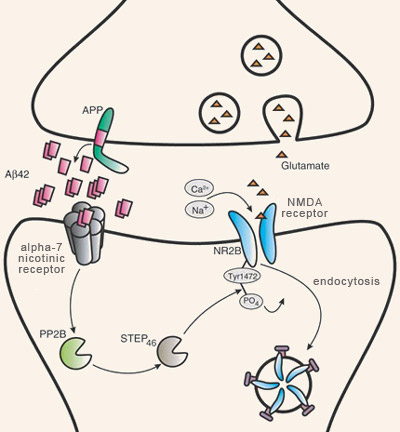
Many scientific observations have shown an association between the aggregation of plaques of beta-amyloid fibrils and the cognitive deficits that characterize Alzheimer’s. However, where and when these amyloid plaques develop in the aging brain is rather poorly correlated with the gradual appearance of the memory losses and other symptoms of Alzheimer’s. Moreover, some cognitively normal individuals develop amyloid plaques without any trace of the neuronal damage associated with them. For this reason, research on the molecular mechanisms of Alzheimer’s has begun to focus less on the non-soluble extracellular deposits of beta-amyloid plaques and more on the soluble, non-fibrillar, oligomeric form of beta-amyloid. In particular, researchers are looking at the role that this form of beta-amyloid may play in synaptic pathology when produced in excessive quantities (in lesser concentrations, it appears to plays a physiological role that is not yet well understood). The synaptic beta-amyloid hypothesis is the name given to this explanatory model for Alzheimer’s in which the central element is the synaptic toxicity of beta-amyloid oligomers, rather than the toxicity of beta-amyloid fibrils agglutinated into plaques. This hypothesis is based on at least two well documented phenomena: a) in humans with Alzheimer’s, the high correlation between the loss of synapses and the clinical severity of their condition, and b) in animal models of Alzheimer’s, the observed rise in soluble beta-amyloid levels in the cortex as signs of pathology increase. Studies using animal models of Alzheimer’s also have made many observations indicating that the synapses might be affected negatively by this increase in beta-amyloid oligomers. For example, physiological concentrations of beta-amyloid dimers and trimers (but not monomers) have been found to induce a gradual loss of synapses in the hippocampus. In other animal studies, using mice, synaptic losses reducing the effectiveness of long- term potentiation (LTP), one of the molecular mechanisms underlying learning and memory, were observed even before any amyloid plaques appeared. Oligomers of beta-amyloid, which have low molecular weights and are non-fibrillar, also can block LTP. These oligomers are also necessary and sufficient to temporarily disrupt learned behaviours.
Prolonged exposure to high levels of beta-amyloid oligomers also causes shrinkage of the dendritic spines—the protrusions from the neurons’ dendrites that form the post-synaptic part of the synapses. This reduction in the density of the dendritic spines is accompanied by a reduction in the level of debrin, a cytoskeletal protein that, along with the actin filament, modulates synaptic plasticity. An interesting detail: this reduction in debrin can be blocked by memantine, a medication prescribed to Alzheimer’s patients under the brand name Namenda, just as the administration of the antibody to beta-amyloid also prevents this deterioration of the dendritic spines.
Around 2009, researchers also established a possible link between the neuronal death typical of Alzheimer’s and a mechanism for eliminating excess neuronal connections that plays a predominant role at the very start of brain development. Their hypothesis was that this mechanism might be reactivated by some processes associated with aging, processes that involved not beta-amyloid itself, but rather the release of the N-terminal fragment, which lies adjacent to beta-amyloid on the APP molecule. This fragment would then trigger a cascade of harmful molecular reactions by binding to a receptor called Death Receptor 6 (DR6). DR6, which is heavily expressed in the parts of the brain affected by Alzheimer’s, was already known to trigger the process called programmed cell death, or apoptosis (follow the Tool Module link to the left). Studies had also shown that blocking the activity of DR6 delayed axonal degeneration in vitro and also caused the redundant synapses to remain in place in certain areas of the mouse brain. These findings suggested that the activation of the DR6 receptor by the N-terminal fragment of APP might reactivate programmed-cell-death mechanisms that are normally active at the very start of brain development. In this model, beta-amyloid plays a complementary role, by degrading the synapses, rather than by killing the cells. Another model proposed at about the same time traces the ultimate cause of Alzheimer’s back beyond amyloid plaques to a disruption of the process of cell division. Thus this model too involves the reactivation, later in life, of a process that normally occurs very early in development—in this case, the differentiation of stem cells into neurons. But mature neurons, which are well differentiated in terms of their dendrites and axons, are clearly no longer suited to cell division. Consequently, the reactivation of the processes that lead to cell division would be fatal to the neurons in the brains of adults who have Alzheimer’s. |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
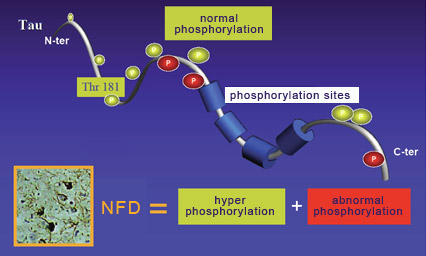
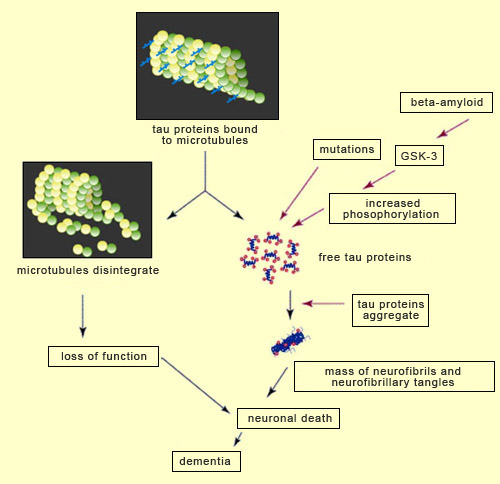
For over a decade, the amyloid-cascade hypothesis dominated research on Alzheimer’s. According to this hypothesis, Alzheimer’s is caused by the accumulation of amyloid plaques (or of beta-amyloid oligomers) that lead to neurofibrillary tangles and then neuronal death. But since the start of the 2000s, numerous findings have begun to cast doubt on this hypothesis and lend more credence to alternative explanations. The best known is probably the one that attributes the primary role in Alzheimer’s pathology to the aggregation of tau proteins to produce neurofibrillary tangles. Without tau protein molecules to stabilize them, the neurons’ microtubules disintegrate, which disrupts axonal transport and ends up killing the neurons. And loss of neurons is very strongly correlated with the seriousness of the cognitive deficits displayed by people with people with Alzheimer’s. As regards the mechanisms by which the tau protein molecules become detached from the microtubules, many scientists believe that increased phosphorylation of the tau protein molecules plays an important role. Indeed, researchers have observed that agglutinated tau proteins are highly phosphorylated and that phosphorylation of tau proteins reduces the strength of their bonds to the microtubules. However, phosphorylation is a complex phenomenon that is involved in the regulation of many processes, including neuronal development. Hence the specific role of phosphorylation in tau protein pathologies (also called “tauopathies") such as Alzheimer’s is still the subject of debate. For example, some data suggest that phosphorylation of tau protein molecules occurs after their aggregation, and that the detachment of these molecules (from the microtubules) and their subsequent aggregation are associated more with structural changes within them than with their phosphorylation. But there is still a great deal of support for the hypothesis that phosphorylation plays a fundamental role in tauopathies. For example, in these pathologies, tau protein molecules develop new phosphorylation sites not found on the tau proteins of healthy developing or adult cells. Note that not all of the known phosphorylation sites on the tau protein molecule are involved in regulating its binding to microtubules. Examples of the sites that do have an influence on this binding are site Thr181 (for the 181st amino acid, which is a threonine) and sites Ser202, 214, 262, 324 and 356 (for the amino acid serine at these respective positions).
The phosphorylation status of tau proteins depends on the balance between the activities of two types of enzymes: protein kinases (which add phosphate groups) and phosphatases (which remove them). Thus, on the one hand, there will be kinases such as protein kinase A, phosphorylase kinase, and glycogen synthase kinase 3, and on the other there will be serine and threonine phosphatases whose antagonistic activities will also help to regulate tau protein activity. However, these enzymatic interactions affecting the phosphorylation of tau protein molecules are very hard to study in vivo, for several reasons. First of all, the activity of kinases in situ often depends on their own phosphorylation status. In other words, the activity of kinases that can phosphorylate the tau protein is itself dependent on the activity of other kinases. These other kinases, which participate in other cascades of biochemical reactions, can thus influence the phosphorylation status of tau proteins indirectly even though they do not interact with them directly. Second, phosphatases can also dephosphorylate and thus deactivate the protein kinases that can phosphorylate tau proteins, so here again, there are possible indirect effects. Moreover, several studies show that there are some co-factors that also can modulate the status of the tau protein by increasing or decreasing its phosphorylation. Examples include the intracellular levels of calcium, cyclical AMP, and phospholipids that influence kinases such as protein kinase A. These co-factors probably alter the three-dimensional structure of tau proteins, possibly making them better substrates for certain kinases. As if all that were not complex enough, the phosphorylation of tau proteins can also by modulated by the physiological state of the cell. When the cell is under stress, phosphorylation of tau proteins increases sharply.
That said, researchers are beginning to take a closer interest in certain specific tau-protein-related enzymes that may be implicated in Alzheimer’s. Many of these enzymes act prior to the biochemical cascade and promote both the aggregation of tau proteins and the formation of amyloid plaques. One such enzyme is glycogen synthase kinase 3 (GSK-3), a protein kinase responsible for the phosphorylation of a number of proteins, including the tau protein. The reason for the interest in GSK-3 is that in addition to its effect on the tau protein, it also regulates the cleavage of APP (the precursor of beta-amyloid), reduces neurogenesis, and increases apoptosis (follow the Tool Module link to the left). All of these phenomena are closely associated with the cognitive deficits seen in Alzheimer’s. Researchers have also discovered some mutations in the gene for the tau protein. These mutations result in tau protein molecules that agglutinate more and bind to microtubules less—another set of conditions conducive to the detachment of tau proteins and their aggregation, resulting in neurofibrillary degeneration. |
| |
|
|
|
|
|
|
|
|