Even at rest, most of your muscles
are in a state of partial contraction called muscle tonus.
This tonus is maintained by the constant activation of a small number of motor
units that contract alternately.
The muscle fibres that make up a
particular motor
unit are not physically located right next to each other, but rather scattered
at various locations in the muscle. This arrangement makes functional sense when
you consider how much energy must be provided to make a muscle contract. This
energy comes from the combustion of glucose, which is carried to the muscle fibres
by the blood capillaries running between them. Since by definition, all the muscle
fibres making up a given motor unit are innervated by the same motor neuron and
contract at the same time, if they were physically adjacent, then when they contracted,
their thickening would compress the capillaries supplying them with energy. This
sudden reduction in their energy supply would produce rapid fatigue in the motor
unit. The scattered distribution of the unit's fibres throughout the muscle avoids
this problem.
THE ROLE OF THE NEUROMUSCULAR JUNCTION IN
MUSCLE CONTRACTION
The contraction of a
motor
unit in a muscle is initiated by the release of acetylcholine into the neuromuscular
junction.
The
acetylcholine activates cholinergic nicotinic
receptors in the motor end plate of the muscle fibre, triggering an excitatory
potential in its post-synaptic membrane, the sarcolemma. If this potential reaches
a certain threshold, a muscular action potential is generated by the potential-dependent
sodium channels in this membrane.
This action potential travels first
over the surface of the sarcolemma and then over that of the T tubules, causing
calcium stored in the sarcoplasmic reticulum to be released. This calcium diffuses
into the myofibrils, which are divided by Z stripes into segments
called sarcomeres. In each sarcomere, thick and thin filaments
then slide past each other, thus drawing the Z stripes closer together, reducing
the length of the sarcomere, and causing the muscle to contract.
To understand how the calcium causes these thick
and thin filaments to slide past each other, we must consider the proteins of
which they are composed. The thick filaments consist mostly of myosin,
while the thin filaments consist mostly of actin. Each myosin
molecule has a "head" at either end, including a site that can bind
with actin.
When no calcium is present, the myosin
in the thick filaments cannot bind with the actin in the thin ones, because the
binding sites on the actin molecules are occupied by another protein, troponin.
But when calcium is released by a muscular action potential, it binds to the troponin,
thereby accomplishing two things: 1) exposing the actin-binding sites on the myosin
molecule heads; and 2) altering the form of another protein, tropomyosin,
so that it exposes the myosin-binding sites on the actin molecules.
The
myosin heads can then bind to the sites on the actin molecules. In this process,
these heads undergo a change in conformation that makes them rotate. It is this
rotation that pulls the thin actin filaments past the thick myosin filaments,
one notch at a time, like a ratchet mechanism, causing the muscle to contract.
The contraction will continue as long as calcium and
ATP are available. One of the functions of the ATP is to break the bond between
the myosin and the actin. (This explains why the muscles of a dead body become
rigid as the supply of ATP begins to run short.)
The
amount of calcium released by the sarcoplasmic reticulum depends on the frequency
of the action potentials in the muscle fibre. (If this frequency reaches or exceeds
50 stimuli per second, it is high enough to cause a sustained muscle contraction,
known as a tetanus.)
The muscle contraction ends when
the action potentials cease and the concentration of calcium in the myofibrils
diminishes. This reduction in calcium is due to its being recaptured by the sarcoplasmic
reticulum, an active process that requires ATP. When the calcium concentration
returns to normal, the muscle fibre returns to its relaxed position.
Myasthenia gravis is a disease associated
with a malfunction in the transmission of impulses from the nerves to the voluntary
muscles. It is considered an auto-immune disease, because in patients who have
it, certain white blood corpuscles manufacture antibodies against the body's own
acetylcholine receptors, thus destroying them. The efficiency of the neuromuscular
junctions is thereby reduced, causing muscle weakness during physical effort or,
in the most serious forms of the illness, a permanent reduction in muscle strength.
Certain toxic gases such
as sarin act by preventing acetylcholinesterase from hydrolyzing acetylcholine,
thus causing constant stimulation of the receptors and violent muscle spasms.
Miniature electrical currents, each
corresponding to the spontaneous release of one acetylcholine (ACh) vesicle, can
be recorded at the postsynaptic motor end plate of the neuromuscular junction
in the absence of any stimulation of the presynaptic motor axon. Each of these
miniature currents corresponds to the opening of about 1 600 nicotinic receptor
channels by the effect of ACh. Since it takes two ACh molecules to open one such
channel, it follows that one vesicle contains 3 200 molecules of ACh. The
spike in the current at the motor end plate represents about 100 miniature currents,
or the release of about 320 000 molecules of ACh opening about 160 000
nicotinic receptors.
NICOTINIC ACETYLCHOLINE RECEPTORS
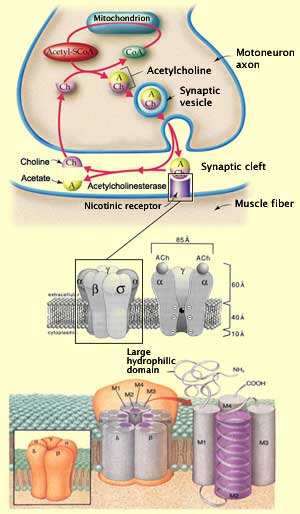
At the neuromuscular
junction, acetylcholine is synthesized in the cytoplasm of the axon's terminal
button, from acetyl coenzyme A and choline. (About half of this choline has been
recaptured by the terminal button after previously produced acetylcholine was
hydrolyzed by the enzyme acetylcholinesterase in the synaptic gap.) Many thousands
of molecules of acetylcholine are thus stored in each synaptic vesicle (see sidebar).
As soon as the vesicles' contents are released
into the synaptic gap, about half of the acetylcholine molecules are hydrolyzed
by acetylcholinesterase. But soon so many acetylcholine molecules accumulate that
this enzyme cannot break them all down, and the remaining half reach the nicotinic
acetylcholine receptors on the postsynaptic side of the gap. Of course, all of
this happens very quickly. Acetylcholinesterase can hydrolyze 4000 molecules of
acetylcholine per site per second, so the half-life of the acetylcholine is estimated
at 1 or 2 milliseconds. In fact, the distribution of the acetylcholinesterase
perfectly matches that of the postsynaptic nicotinic receptors.
Thus,
in the case of acetycholine, synaptic
transmission is terminated chiefly by a biochemical reaction. In contrast,
for catecholamines, the main mechanisms are diffusion of the neurotransmitter
outside the synaptic gap and, primarily, its recapture by the presynaptic button.
Acetylcholine receptors are divided into two major families: nicotinic
and muscarinic. The acetylcholine receptors in neuromuscular junctions are
nicotinic. They consist of pentameric proteins that form an ion channel embedded
in a lipid bilayer in the postsynaptic membrane. These receptors number nearly
10 000/µm2. The acetylcholine binds to the extracellular part
of the two alpha subunits of this channel protein, which has 5
subunits in all.
Each subunit in turn consists of four helicoidal
transmembrane domains, designated M1 to M4. It is the M2 domains of the five subunits
that together form the wall of the ion channel. This ion channel, which is opened
by the allosteric conformation change triggered by the binding of the acetylcholine,
is equally permeable to sodium and potassium. Its permeability to calcium accounts
for only 2.5% of its total permeability. The half-life of adult nicotinic receptors
is 4 to 6 days.