Corticosteroids (also
known as corticoids) are hormones secreted by the outer portions of the adrenal
glands. They can be divided into three groups, for each of which there are separate
receptors: androgens, which are involved in the development of
sexual traits; mineralocorticoids (aldosterone, corticosterone,
desoxycortisone), which regulate the body's osmotic balance; and glucocorticoids
(cortisone, hydrocortisone, prednisone), which, in addition to their
anti-inflammatory and immunosuppressive effects, stimulate the synthesis of glucose
and increase the mobilization of fatty acids and proteins to meet the higher metabolic
demands generated by stress.
Glucocorticoids play an
extremely important role in fear and anxiety reactions and in depressive states.
These hormones often affect behaviour by increasing or decreasing the efficiency
of certain neural pathways.
SEROTONIN AND OTHER MOLECULES INVOLVED IN
DEPRESSION
When someone perceives
a situation as disagreeable or dangerous, a general response to this stress is
triggered in their body. Depending on the situation and the person's experience
with such situations, he or she will choose
a behaviour: either fight,
or flight, or inhibition of action (the status quo).
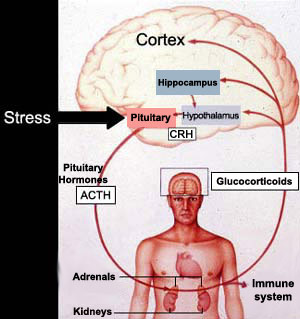
The body's response
from the time it perceives a danger to the time it secretes the hormones to prepare
to deal with it involves the following structures, in the following order: 1)
the limbic system, 2) the hypothalamus, 3) the pituitary gland, and 4) the adrenal
glands. The adrenal glands secrete glucocorticoids (such as cortisol, in human
beings), which interact with the serotonin receptors in the brain.
When someone experiences a stressful
event, the level of glucocorticoids in their blood rises.
Stress activates the hypothalamus, which then secretes corticotropin-releasing
hormone (CRH). The CRH in turn causes the pituitary gland
to release adrenocorticotropic hormone (ACTH) into the bloodstream,
from which it enters the adrenal glands and causes them to
secrete cortisol.
This process creates a negative feedback loop in which the excess cortisol
activates the brain's glucocorticoid receptors and suppresses the production of
CRH. In depressed patients, however, this loop no longer works, resulting in excess
production of CRH and hence of cortisol. Many seriously depressed patients have high blood levels of cortisol, caused
by chronic stress.
In rats, chronic
stress and/or a high level of glucocorticoids alters certain serotonergic receptors
(increases the 5-HT2Areceptors
in the cerebral cortex and reduces the 5-HT1Areceptors in the hippocampus). These same changes have been observed in
humans who have committed suicide
or suffered from diseases that cause hypersecretion of glucocorticoids. The continued
administration of antidepressants causes changes in the serotonergic receptors
that are the opposite of the changes produced by chronic stress. It also reverses
the hypersecretion of stress hormones.
Not incidentally, in humans, many
glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs) (see sidebar)
are located in the hypothalamus and the hippocampus, two structures involved in
mood control and the ability to experience pleasure. These receptors are sensitive
both to the levels of the various corticosteroids in the body and to the length
of time that they are active, so their activation mechanisms will have direct
impacts on the behavioural response chosen to a given stimulus.
Prolonged chronic stress also seems
to alter the response of the MR and GR receptors and to have very harmful effects
on people's mental equilibrium, especially when social or family supports are
absent. Under these conditions, the glucocorticoid response, which was originally
highly adaptive, becomes clearly maladaptive.
It has long been known that depressed
persons display hyperactivity in the hypothalamic-pituitary-adrenal (HPA) axis
(see illustration and explanation above). A prolonged state of inhibition
of action is also known to encourage the emergence of a depressive state.
This chronic excess stress on the HPA axis is believed to result in structural
changes in certain parts of the brain. For example, region
CA3 of the hippocampus loses large numbers of neurons when subjected to
prolonged stress.
Other studies have also reported a reduced number
of glucocorticoid receptors in the hippocampus and prefrontal cortex of suicide
victims. Though it is hard to tell whether these structural changes are of genetic
origin or the result of chronic activation of the HPA axis, they would be consistent
with hyperactivity in this axis when the natural braking effect of these receptors
was reduced.
Here's another example: people with Cushing's syndrome,
a disease in which the body produces excess cortisol, display a high incidence
of depression, and their depression lifts when they are given treatments that
bring their cortisol levels back to normal.
Thus, all indications are
that the end products of the HPA axis—glucocorticoids— play a role
in depression by influencing several neurotransmitter systems, including those
for serotonin, norepinephrine, and dopamine, all three of which are involved in
depression.
Treatment with antidepressants is
often regarded as consisting of two phases. During the first two weeks, the patient's
depressive state does not really improve. But after two or three weeks, the patient
gradually begins sleeping and eating again, feeling more energetic, and having
more positive thoughts. It is recommended that treatment then be continued for
several months to minimize the risk of a relapse.
Various hypotheses
have been offered to explain this lag before antidepressants become effective.
One hypothesis is that at the start of the treatment, once the antidepressant
medication has inhibited the reuptake of serotonin, the serotonin autoreceptors
quickly become saturated, so that their inhibitory effect predominates, reducing
the amount of serotonin released into the synaptic gap. But over time, these autoreceptors
become desensitized, and the presynaptic neuron can produce action potentials
more readily. Because the antidepressants are still preventing the serotonin from
being reabsorbed, its extracellular concentration increases, and serotonergic
transmission is facilitated.
The effect of antidepressants can
be compared with that of ecstasy,
which causes the release of large amounts of serotonin from the nerve endings
of neurons. This excess of serotonin is suspected to be the source of the particular
mental effects of ecstasy, including those associated with feelings of well-being—effects
analogous to those of antidepressants.
An initial hypothesis formulated in the 1960s
identified norepinephrine as the main neurotransmitter involved
in depression. According to this “catecholamine hypothesis”, depression
was due to a deficiency of norepinephrine in certain circuits of the brain, while
mania was due to an overabundance of this same neurotransmitter. Though this hypothesis
is still recognized, it does not explain everything, in particular why there are
some people whose mood is not affected by fluctuations in their norepinephrine
levels.
In the 1970s, the “permissive hypothesis” emerged,
which postulated that another neurotransmitter, serotonin, was
involved in depression. According to this hypothesis, a reduction in the amount
of serotonin in certain synapses may cause depression by triggering or “permitting”
a drop in norepinephrine. Consequently, though norepinephrine was still acknowledged
to play an important role in depression, attempts could now be made to treat depression
by acting on the body's serotonin levels. This has been the therapeutic approach
applied using Prozac and all the other selective serotonin reuptake inhibitors
(SSRIs) that have come on the market since the 1980s.
Fluoxetine (Prozac)
A
third neurotransmitter of importance in depression is dopamine,
the same molecule that is involved in schizophrenia and Parkinson's disease. Dopamine
plays an important role in rewards and positive reinforcement— in other
words, in the pursuit of gratification. The use of dopaminergic substances and
stimulants as antidepressants produces quick, positive results in many patients,
which makes these substances useful complements to other antidepressants that
may take several weeks to act.
Medications that act directly on dopamine
must be used cautiously, however, because they can create dependencies. Many drugs,
such as cocaine, opiates, and alcohol, increase the production of dopamine, which
may explain why many people with depression use them.
Many researchers now believe that
it is not really appropriate to describe the physiological causes of depression
as a "chemical imbalance".
The hypothesis
that depression was caused by a "chemical imbalance" originated in the
1960s. The first antidepressant medications, developed around that time, were
tricyclics and monoamine oxidase inhibitors (MAOIs). In addition to alleviating
the symptoms of depression in many patients, these molecules were known to increase
brain levels of dopamine, norepinephrine, and serotonin in various ways. For this
reason, researchers hypothesized that depression might be due to an imbalance
in these neurotransmitters. This hypothesis did indeed yield some fairly useful
research findings during the last decades of the 20th century. In addition, by
emphasizing that mood disorders might be related to a physiological malfunction
and not simply to a character defect or a lack of will power, this hypothesis
reduced the needless feelings of guilt that often haunted people with depression.
However,
the results of efforts to identify this "chemical imbalance" more precisely
have been rather disappointing and contradictory. Research is now focusing less
on the neurotransmitters themselves and more on the receptors for these molecules
and on the molecular events involved in regulating genes. But here too, there
is room for controversy. Relatively little direct evidence has been found of alterations
in receptors or anomalies in gene expression related to these receptors or other
enzymes in cases of depression. Moreover, the reason for the two to three week
lag between the time when antidepresssants first affect neurotransmitters and
the time when they begin to affect mood (see sidebar to the left) is still not
well understood. In short, the situation is far more complex than scientists believed
in the 1960s when they first formulated the "chemical imbalance" hypothesis.
Given
these problems in securing any unequivocal data to support this hypothesis, some
scientists have begun to ask whether the extensive use that continues to be made
of the term "chemical imbalance" might raise some ethical problems,
or even political ones. In the United States, for example, where advertisements
for antidepressants are allowed in the
mass media, the pharmaceutical companies have not always worried
about this fine point. Simplistic advertisements tell Americans that when they
are depressed, a substance in their brain is out of balance, and that if they
take the right antidepressants, the ideal balance will be magically restored.
These advertisements may well have something to do with the stunning success of
SSRI antidepressants, such as Prozac, Zoloft, and Paxil, in the marketplace, and
the billions of dollars that they have earned for the companies that make them.
The effects of antidepressants are
not limited to the presynaptic neurons. In postsynaptic neurons, the antidepressive
effect of tricyclics and MAOIs may be attributable to “down-regulation”
(reduction in the number but not the sensitivity) of the beta-adrenergic receptors
and the 5-HT2 serotonergic receptors. Desensitization is also observed
in the norepinephrinergic receptors coupled to adenylate cyclase . The phenomena
of transduction via the G-proteins paired with the receptors represent another
possible site of postsynaptic effects, as is probably the case for
lithium.